- 在線諮詢
- 返回頂部
歡迎光臨長泰化學工業 ( 惠州 ) 有限公司官方網站!

+86-752-687-3099


NEWS
公司資訊

安徽師範大學代勝瑜教授課題組J. Catal.文章:金屬-π相互作用下雜原子二苯並環庚基對乙烯聚合鏈轉移與鏈行走的調控研究
鏈行走與鏈轉移是決定通過後過渡金屬催化乙烯(共)聚合所生產聚烯烴結構和分子量的兩大核心因素。這些過程受多重因素影響,既包括外部聚合條件,例如助催化劑類型、溫度、單體濃度及鏈轉移試劑,也涵蓋催化活化位點周邊的配體環境,特別是配體的空間位元阻和電子效應。在諸多因素中,配體空間位元阻在後過渡金屬催化體系中起到了關鍵的調節作用,用以平衡鏈行走、鏈轉移及鏈增長之間的關係。研究顯示,在多數後過渡金屬催化體系中,增大催化中心的軸向空間位阻能有效抑制鏈轉移,進而在相同條件下生產出分子量更高的聚合物。例如,一系列不對稱二芳基甲基α-二亞胺鎳和鈀催化劑(圖1a-b)的空間位阻逐步增加時,所得聚乙烯和共聚物的分子量也隨之逐步增加。然而,這種趨勢並非放之四海而皆準,因為鏈增長同樣可能受到空間位阻的制約。在某些情境下,鏈增長受到的抑制可能更為顯著,導致所得聚合物的分子量反而低於那些由弱遮罩催化中心生產的聚合物。例如,某些不對稱α-二亞胺鎳催化劑能迅速產出高分子量聚乙烯。同樣地,空間位阻的變化也可能影響鏈行走過程及所得聚合物的微觀結構,諸如支化度的改變。先前的研究揭示,隨著鈀催化中心空間位阻的增加,所得聚乙烯和共聚物的支化度逐漸降低(Macromolecules 2016, 49, 8855–8862)。相反地,在相應的鎳催化體系中,聚乙烯的支化度最初隨鎳催化中心空間位阻的增加而上升,隨後又出現下降(Organometallics 2019, 38, 2919-2926)。這些發現凸顯了通過調整配體空間位阻,我們能夠定制出具有理想分子量和支化度的聚合物。經過精心調控,我們可以改變聚烯烴的結構與性能,從而製備出目標聚合物。

此外,調節配體的電子效應也有可能在一定程度上影響所得聚合物的分子量、支化度和拓撲結構,這一點在中性水楊醛亞胺鎳體系中尤為明顯。例如,Mecking等人報導的N-三苯基水楊醛亞胺鎳催化劑中,具備吸電取代基(如CF3、SF5、NO2)的催化劑能聚合得到高分子量線性聚合物,而具備給電子取代基(如Me、OMe)的催化劑則在相同條件下聚合得到超支化低分子量聚合物(圖1c)(Acc. Chem. Res. 2020, 53, 2738–2752)。理論計算與實驗證據均表明,鄰芳基芳環與鎳中心之間較弱的相鄰基團相互作用有利於β-H的消除,進而促進鏈轉移與支化形成。近期的一些報導還發現了類似的弱鄰基相互作用,涉及[N^N]體系中陽離子鎳和鈀的遠端非共軛電子效應(圖1d)( Polym. Chem. 2020, 11, 2692–2699)及第二層配位空間效應(圖1e)( Angew. Chem., Int. Ed. 2017, 56, 11604–11609)。儘管已觀察到一些有意義的模式,但這些相互作用對聚合活性、分子量及所得聚合物微觀結構的影響尚不十分明確。在本研究中,我們通過用具有不同給電子能力的雜原子取代催化劑中橋接的亞乙基碳原子,從而實現了對所得(共)聚合物分子量的精確調控(圖1f)。值得注意的是,我們觀察到弱金屬-π相互作用實際上是抑制而非促進鏈行走,這與先前報導的趨勢形成了鮮明對比。

圖1. 配體空間位元阻(a-b)和電子效應(c-f)修飾的後過渡金屬催化劑

圖2. 含二苯並環庚基及雜原子取代的二苯並環庚基α-二亞胺配體和其相應的鎳和鈀催化劑

鎳和鈀催化劑的合成
按照先前報導的方法,我們採用NaBH4還原對應的二苯並庚酮,成功合成了二苯並庚醇及其雜原子取代的醇類(O1-O3)。緊接著,我們通過金屬催化的Friedel-Crafts烷基化反應,進一步製備了單側苯並環庚基苯胺及其含雜原子的取代胺類(A1-A3)。之後,在對甲苯磺酸的催化作用下,我們將苯胺A1-A3與2,3-丁二酮在120 ℃的甲苯環境中進行縮合,從而得到了α-二亞胺配體L1-L3(詳見圖2)。為了確保化合物的準確性,我們使用了1H和13C NMR以及高解析度質譜(HRMS)對所有新合成的化合物進行了詳盡的表徵。當α-二亞胺配體(L1-L3)與1當量的(DME)NiBr2或(COD)PdMeCl反應時,能夠生成相應的催化劑Ni1-Ni3和Pd1-Pd3,且產率分別達到了59-74%和52-70%(參見圖2)。我們對鎳催化劑進行了元素分析表徵,同時,對鈀催化劑則採用了更為全面的1H和13C NMR、元素分析以及X射線單晶衍射進行表徵。為了進一步探究催化劑的結構,我們通過乙醚對催化劑的二氯甲烷溶液進行分層,成功獲得了催化劑Pd1和Pd2的單晶,並對其進行了X射線衍射分析。分析結果顯示,這兩種鈀催化劑在Pd金屬中心周圍均呈現出平面正方形幾何結構(見圖3)。從單晶結構中我們可以清晰地觀察到,二苯並環庚基取代基對鈀中心的軸向位置起到了有效的遮罩作用。特別是在催化劑Pd2的單晶中,兩個氧原子與金屬中心之間的距離(Pd1-O1=6.31Å,Pd1-O2=6.87Å)遠大於兩個原子間的范德華半徑總和(3.15Å),這表明氧原子與金屬中心之間直接相互作用的可能性極低。相較之下,二苯並環庚基取代基中的苯基中心與金屬中心的距離僅為3.95 Å,這揭示了在聚合過程中可能存在弱相互作用,因為這個距離已接近金屬-π相互作用的臨界值。此外,系統中的雜原子能夠通過調節苯環上的電子密度來影響與金屬中心的相互作用強度,這種調節機制為控制乙烯聚合過程中的鏈轉移和鏈行走提供了有效手段。鎳催化乙烯聚合經過200當量助催化劑Et2AlCl的活化處理後,三種鎳催化劑在乙烯聚合反應中均展現出了卓越的活性。在相同的實驗條件下,這三種催化劑的活性排序為Ni1>Ni2>Ni3,其中Ni1的活性顯著高於其他兩種催化劑(見圖4a)。Ni2和Ni3活性的降低,可能是由於雜原子取代的二苯並環庚基與催化中心之間產生了更強的金屬-π相互作用,這種作用抑制了鏈的增長過程(見圖5a)。特別值得注意的是,含有硫的取代基對催化劑活性的抑制作用更為顯著。隨著溫度從30℃升高到50℃,Ni1和Ni3的聚合活性逐漸增強,但在70℃時活性開始下降。與此不同,Ni2的聚合活性則隨著溫度的升高而持續下降(見圖4a)。這種變化可以歸因於催化劑穩定性和活性隨溫度變化的相互作用。金屬-π相互作用不僅對催化劑的活性有影響,還顯著改變了所得聚乙烯的分子量和微觀結構。由Ni1催化制得的聚乙烯,其分子量明顯高於由其他兩種催化劑制得的聚乙烯,差距超過一個數量級(見圖4b)。在乙烯聚合過程中,富電子的雜原子取代二苯並環庚基通過與雙鍵中間體競爭配位元到金屬中心,從而促進了鏈轉移過程。這種促進作用隨著取代基電子豐度的增加而增強(見圖4b和圖5b)。聚乙烯的分子量主要由鏈增長速率與鏈轉移速率的比值決定。由於金屬-π相互作用在加速鏈轉移的同時減緩了鏈增長,因此這一比值顯著降低,導致聚乙烯的分子量大幅下降。同時,隨著溫度的升高,所得聚乙烯的分子量逐漸降低(見圖4b),這一觀察結果與先前關於α-二亞胺鎳體系的報導相一致,即溫度升高會導致鏈轉移與鏈增長的比率上升。最引人注目的是,這種金屬-π相互作用對聚乙烯的支化度也產生了顯著影響。如圖4c所示,與Ni2(43-48/1000C)和Ni3(21-32/1000C)催化合成的聚乙烯相比,Ni1(83-90/1000C)催化合成的聚乙烯具有明顯更高的支化度。富電子的雜原子取代二苯並環庚基通過抑制乙烯聚合過程中的β-H消除來阻礙鏈行走,從而降低聚乙烯的支化度(見圖6)。特別是更富電子的硫代二苯並環庚基取代基,對鏈行走的抑制作用更為顯著,導致聚乙烯的支化度最低(見圖4c和圖6)。此外,根據先前對α-二亞胺鎳體系的研究,溫度升高會增加聚乙烯的支化度,因為高溫條件更有利於鏈行走而非鏈增長。這些支化聚乙烯的熔點各不相同,許多樣品甚至表現出雙熔點特性。這種現象可能是由多種因素共同作用的結果,如聚合物分子量的降低以及非均質支化結構等,這些因素都可能導致聚合物的結晶過程變得不規則。
圖4. 在30-70 ℃條件下,Ni1-Ni3生成聚乙烯的產量(a)、分子量(b)和支化度(c)

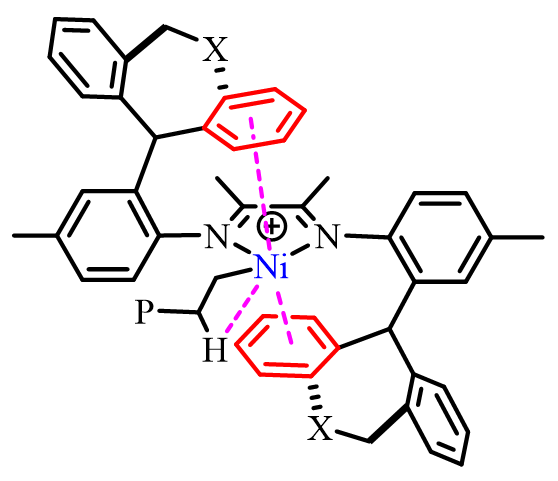
圖6. 富電子的雜原子二苯並環庚基與β-H競爭與金屬中心的相互作用抑制β-H消除的反應機制
鈀催化乙烯(共)聚合經過2.0當量的四(3,5-二(三氟甲基)苯基)硼酸鈉(NaBArF)的原位活化後,鈀催化劑展現出了中等活性。在30℃時,裝載了含雜原子二苯並環庚基取代基的催化劑Pd2和Pd3的活性超過了Pd1。然而,當溫度升高到50℃時,這一活性趨勢發生了反轉(圖7a)。與鎳體系相比,鈀體系中的反應情況更為複雜,這種複雜性可能源自多種因素,例如Pd金屬中心對富電子芳基的更高耐受性以及溫度對這些相互作用的複雜影響。與鎳體系的觀察結果相呼應,Pd2在所有測試溫度下均優於Pd3,尤其是當取代基為含硫的更富電子基團時,鏈增長受到的抑制更為明顯(見圖7a)。值得注意的是,隨著溫度的升高,所有鈀催化劑的活性普遍增強,其中Pd1的活性增幅最為顯著。這一提升很可能是由於在較高溫度下,乙烯插入的能量壁壘有所降低(如圖7a所示)。與鎳體系的結果相類似,鈀體系中的金屬-π相互作用對所生成的聚乙烯的分子量產生了顯著影響。鈀催化乙烯聚合所得聚乙烯的分子量遵循Pd1>Pd2>Pd3的順序(如圖7b所示)。值得注意的是,由Pd3催化得到的聚乙烯分子量明顯低於其他兩種催化劑,差距超過一個數量級(請見圖7b)。這一發現進一步印證了含硫二苯並環庚基取代基對鈀催化中心的深遠影響。與鎳體系相一致的是,隨著溫度的升高,所有催化劑產生的聚乙烯分子量均有所下降,這主要歸因於鏈轉移與鏈增長比率的降低。然而,與鎳體系不同的是,在鈀體系中,僅有含硫的二苯並環庚基取代基對降低聚乙烯的支化度具有顯著影響(請見圖7c)。這可能是因為柔性的、富電子的含硫二苯並環庚基取代基與鈀催化中心之間的相互作用更為有效。對聚乙烯進行13C NMR譜分析揭示了其支化結構主要由長鏈支化構成(如圖8所示),從而驗證了這些聚合物在室溫下於THF中的溶解度。綜上所述,在鈀體系中,含氧的二苯並環庚基取代基在促進鏈轉移和抑制鏈行走方面效果不佳,這可以歸因於鈀中心對富電子環境具有一定的耐受性。
圖7. 30 ℃和50 ℃下,Pd1-Pd3產生聚乙烯的產量(a)、分子量(b)和支化度(c)比較

圖8. 在30 ℃下,Pd2產生的支化聚乙烯的13C NMR譜圖分析
丙烯酸甲酯,作為一種廣泛使用的單體,在乙烯共聚反應中佔據著舉足輕重的地位。它常被用作評估α-二亞胺鈀催化劑共聚性能的基準物質。同時,丙烯酸甲酯在極性官能化聚乙烯的合成過程中也扮演著重要角色。本研究發現,與均聚活性相比,鈀催化劑在共聚反應中的活性顯著降低。這一現象主要是由於丙烯酸甲酯插入後形成了穩定的環狀中間體。與鈀催化的乙烯均聚反應類似,通過Pd3催化的乙烯與丙烯酸甲酯的共聚反應所生成的乙烯-丙烯酸甲酯共聚物,其分子量明顯低於由Pd1和Pd2催化得到的共聚物,同時其支化度也較低(見圖9)。這些觀察結果表明,在乙烯-丙烯酸甲酯共聚過程中,含硫的二苯並環庚基取代基同樣起到了抑制鏈行走並促進鏈轉移的作用。然而,一個引人注目的現象是,Pd3的共聚活性與Pd1相當,且顯著高於Pd2(參見圖9a)。儘管其根本原因仍有待深入探究,但這可能與含硫二苯並環庚基取代基在促進穩定環中間體開環方面的作用有關。值得注意的是,在多數情況下,由Pd1和Pd2催化的共聚物中,丙烯酸甲酯的插入比例超過了Pd3。這一差異的具體原因目前尚不明確,但可能與二苯並環庚基取代基和丙烯酸甲酯單體之間的Pd-π相互作用的特性密切相關。

圖4.9 在1.0和2.0 mol/L丙烯酸甲酯濃度下,用Pd1-Pd3生成的共聚物的產量(a)、插入比(b)、分子量(c)和支化度(d)的比較
綜上所述,本研究利用α-二亞胺配體,成功設計並合成了一系列具有單邊雜原子二苯並環庚基取代基的鎳和鈀催化劑。在鎳催化的乙烯聚合反應中,我們發現雜原子二苯並環庚基鎳催化劑的活性較低,所生成的聚乙烯相較於二苯並環庚基鎳催化劑,具有更低的分子量和支化度。特別值得一提的是,含硫取代的二苯並環庚基鎳催化劑的活性更低,且其產生的聚乙烯分子量和支化度均低於含氧取代的二苯並環庚基鎳催化劑。在鈀催化的乙烯聚合中,我們也觀察到類似的規律:雜原子的二苯並環庚基鈀催化劑相較於對應的二苯並環庚基鈀催化劑,更傾向于生成低分子量和低支化度的聚乙烯。尤為引人注目的是,在鈀體系中,含硫的二苯並環庚基取代基對聚合反應的影響更為顯著。在雜原子二苯並環庚基鈀催化劑的共聚反應中,也呈現出類似的趨勢,顯著特點是能夠生成低分子量且低支化度的共聚物。我們將這些觀察結果歸因於催化中心上相鄰基團間的金屬-π相互作用。具體來說,在乙烯的(共)聚合過程中,與富電子的雜原子二苯並環庚基取代基相關的增強的金屬-π相互作用,有助於促進鏈轉移並抑制鏈行走。特別是在鎳和鈀體系中,含硫的二苯並環庚基取代基因其強給電子能力和較柔性的電子結構,顯著地促進了鏈轉移並抑制了鏈行走。本研究不僅為金屬催化乙烯聚合體系的設計和優化提供了寶貴的參考,更展示了通過合理選擇配體和取代基來精准調節聚乙烯性能的可行性。該研究成果發表在《J. Catal.》(DOI:10.1016/j.jcat.2024.115550),陸衛青和丁北航為該論文共同第一作者。
來源:高分子科學前沿
免責聲明:本內容來自騰訊平臺創作者,不代表騰訊新聞或騰訊網的觀點和立場。